【前沿速递】中国科学家揭示流感病毒抗原变异的决定性位点突变规律
2019年3月9日,国际期刊Molecular Biology and Evolution在线发表了苏州系统医学研究所吴爱平课题组的最新研究成果“Cluster-transition determining sites underlying the antigenic evolution ofseasonal influenza viruses”。该研究揭示了关于两种主要威胁人类健康的季节性流感病毒(甲型H1N1和H3N2)发生抗原变异的决定性位点突变的支配规律,将为流感病毒的变异监测预警和疫苗株选择提供重要指导。

流感病毒的频繁突变,使得病毒能够快速地逃避宿主的免疫压力,导致毒株间显著的抗原变异、流感的季节性流行以及疫苗保护的失效等。而季节性流感A(H1N1)和A(H3N2)的反复变异和流行,给人类社会带来了巨大的公共卫生压力和疾病负担。因此,对于季节性流感病毒抗原变异规律的研究,不仅有助于我们深入掌握病毒的进化变异趋势,还能直接指导流感疫苗株的选取,具有重要的理论意义和实际价值。
为了识别出流感病毒抗原变异(即抗原类替换)背后的关键性位点突变,吴爱平课题组开发了一个基于机器学习的抗原决定性位点识别方法RECDS,通过整合病毒序列、结构和进化信息,系统地度量了流感病毒主要抗原蛋白HA上每个位点在整个流感病毒进化历史中的抗原贡献,识别出了季节性流感A(H3N2)和A(H1N1)抗原变异过程中最关键的10-15个决定性突变位点。
研究发现,流感病毒A(H3N2)的抗原类替换决定性位点主要分布在HA蛋白的受体结合区(RBD)附近,而A(H1N1)除了分布在RBD区外,还分布在另外一个残留酯酶区域(ED),这表明两种亚型病毒既采用了一致的突变策略,又有自身特异性的变异机制。这也一定程度上给出了为什么甲型H3N2病毒抗原变异快而甲型H1N1病毒抗原变异相对较慢的原因。
除此之外,该研究进一步通过结合病毒进化信息,从位点角度刻画出了每个关键位点在抗原变异过程中的氨基酸突变的特征和历史,并从病毒角度描绘出了抗原变异过程中的整体关键位点突变轨迹和支配规律。上述研究结果一方面深化了对于流感病毒变异规律的机制认识,另一方面为病毒变异监测和疫苗株推荐提供了关键位点靶标。
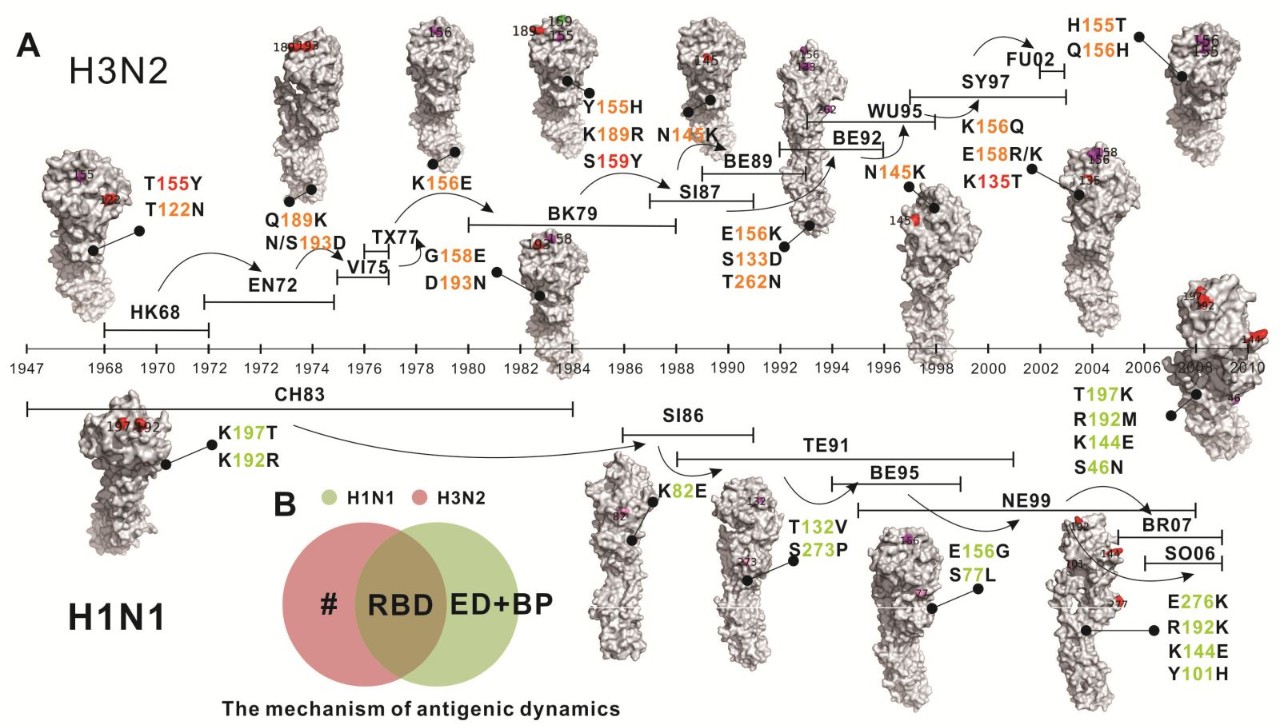
图:流感病毒抗原变异支配性位点动态突变路径
中国医学科学院苏州系统医学研究所吴爱平课题组的权丽君博士为该项工作的第一作者,吴爱平研究员为通讯作者。该研究得到了国家重点研发计划、国防科技创新特区项目、医科院医学与健康科技创新工程、国家和江苏省自然科学基金以及江苏省六大人才高峰等项目的共同支持。
吴爱平课题组
吴爱平课题组是以吴爱平研究员为负责人的传染病生物信息学团队。课题组成立于2016年,迄今已有5名工作人员、1名博士后和研究生3人,成员背景覆盖了机器学习建模、病毒学、免疫学、基因组学和生物信息学等多个互补学科。课题组致力于发展新的计算方法和系统整合模型,研究传染病(如寨卡和流感等病毒感染性疾病)的病原进化变异规律及宿主免疫反应模式等。课题组成员秉持着严谨与创新、合作与交流的科研理念,通过整合病毒序列、结构、功能以及流行学数据等多层次信息,在病毒的进化变异规律上有系统性发现,在宿主免疫定量建模方面取得了持续性进展,并在领域相关的期刊上发表了多篇代表性论文。
ABSTRACT:
Seasonal influenza viruses undergo frequentmutations on their surface hemagglutinin (HA) proteins to escape the hostimmune response. In these mutations, a few key amino acid sites are associatedwith significant antigenic cluster transitions. To recognize thecluster-transition determining sites of seasonal influenza A/H3N2 and A/H1N1viruses systematically and quickly, we developed a computational model namedRECDS to evaluate the contribution of a specific amino acid site on the HAprotein in the whole history of antigenic evolution. In RECDS, we ranked all ofthe HA sites by calculating the contribution scores derived from the forest ofgradient boosting classifiers trained by various sequence-based andstructure-based features. With the RECDS model, we found out that the sitesdetermining influenza antigenicity were mostly around the receptor-bindingdomain both for the influenza A/H3N2 and A/H1N1 viruses. Specifically, half ofthe cluster-transition determining sites of the influenza A/H1N1 virus werelocated in the vestigial esterase domain and basic path area on the HA, whichindicated that the differential driving force of the antigenic evolution of theA/H1N1 virus refers to the A/H3N2 virus. Beyond that, the footprints ofsubstitutions responsible for antigenic evolution were inferred according tothe phylogenetic trees for the cluster-transition determining sites. Themonitoring of genetic variation occurring at these cluster-transitiondetermining sites in circulating influenza viruses on a large scale willpotentially reduce current assay workloads in influenza surveillance and theselection of new influenza vaccine strains.
原文链接:
1. Lijun Quan, Chengyang Ji, Xiao Ding, Yousong Peng, MiLiu,Jiya Sun,Taijiao Jiang, Aiping Wu. Cluster-transition determining sitesunderlying the antigenic evolution of seasonal influenza viruses. MolecularBiology and Evolution, msz050, https://doi.org/10.1093/molbev/msz050
2.苏州系统医学研究苏:吴爱平课题组揭示流感病毒抗原变异的决定性位点突变规律
https://www.ismsz.cn/Index/Page/77a7ceeb-3038-4fdd-bb7c-ddba6249ffdf
内容来源:病毒学界,版权归原作者所有,如有侵权,请联系删除
卓诚惠生——追求品质卓越,致力健康事业
了解更多资讯,请识别下方二维码,关注卓诚惠生~
微信:
ABTechnology


Customer service consultation:+ 86-10-8193-7740
Contact Email:christian@x-abt.com
working hours:9:00-17:30

FOCUS ON US